Lecture 10
Cluster Analysis
To Scale or not to Scale?
Show code
monster_scale_compare |>
ggplot(aes(x = value, y = feature, fill = scale_state)) +
geom_vline(
data = tibble(scale_state = "Z-score scaled", xint = 0),
aes(xintercept = xint),
inherit.aes = FALSE,
linewidth = 0.7,
linetype = "dashed",
colour = "#8a3324"
) +
geom_boxplot(outlier.alpha = 0.2, width = 0.7, orientation = "y") +
facet_wrap(~ scale_state, ncol = 1, scales = "free_x") +
guides(fill = "none") +
labs(x = "Raw units / z-score", y = NULL)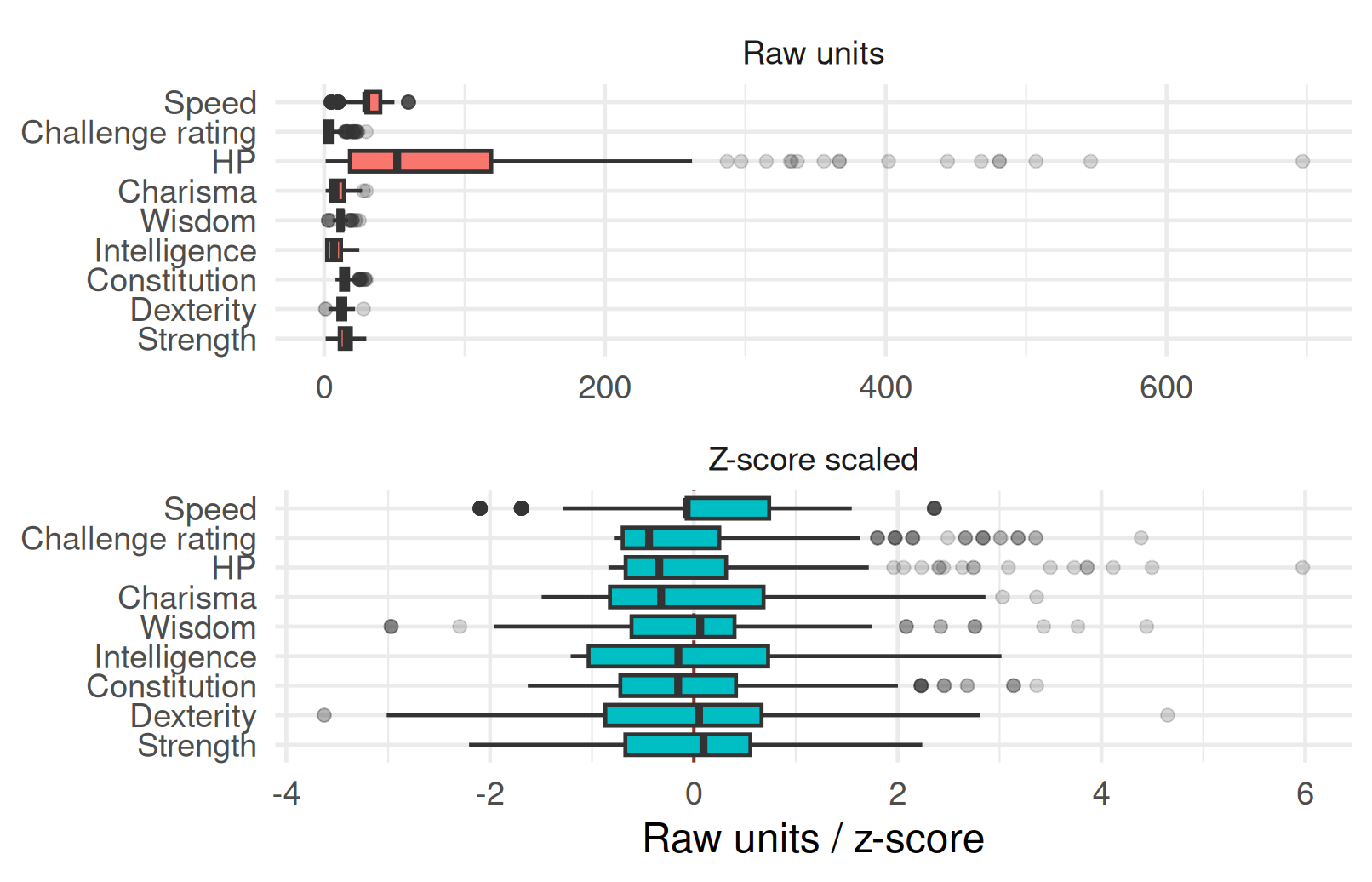
- The D&D ability scores live on a small scale, while
hp_number,cr, andspeed_base_numberare much larger in raw units. - If we cluster on raw Euclidean distance, the large-scale variables dominate the smaller ones.
- So the clustering examples below use z-score standardization before computing Euclidean distances.
- Here
scale()means subtract the column mean and divide by the column standard deviation, not min-max scaling.
Hierarchical clustering on monsters
- The result is a nested tree of clusters, called a dendrogram.
- The tree can be cut at different heights to give different clusterings.
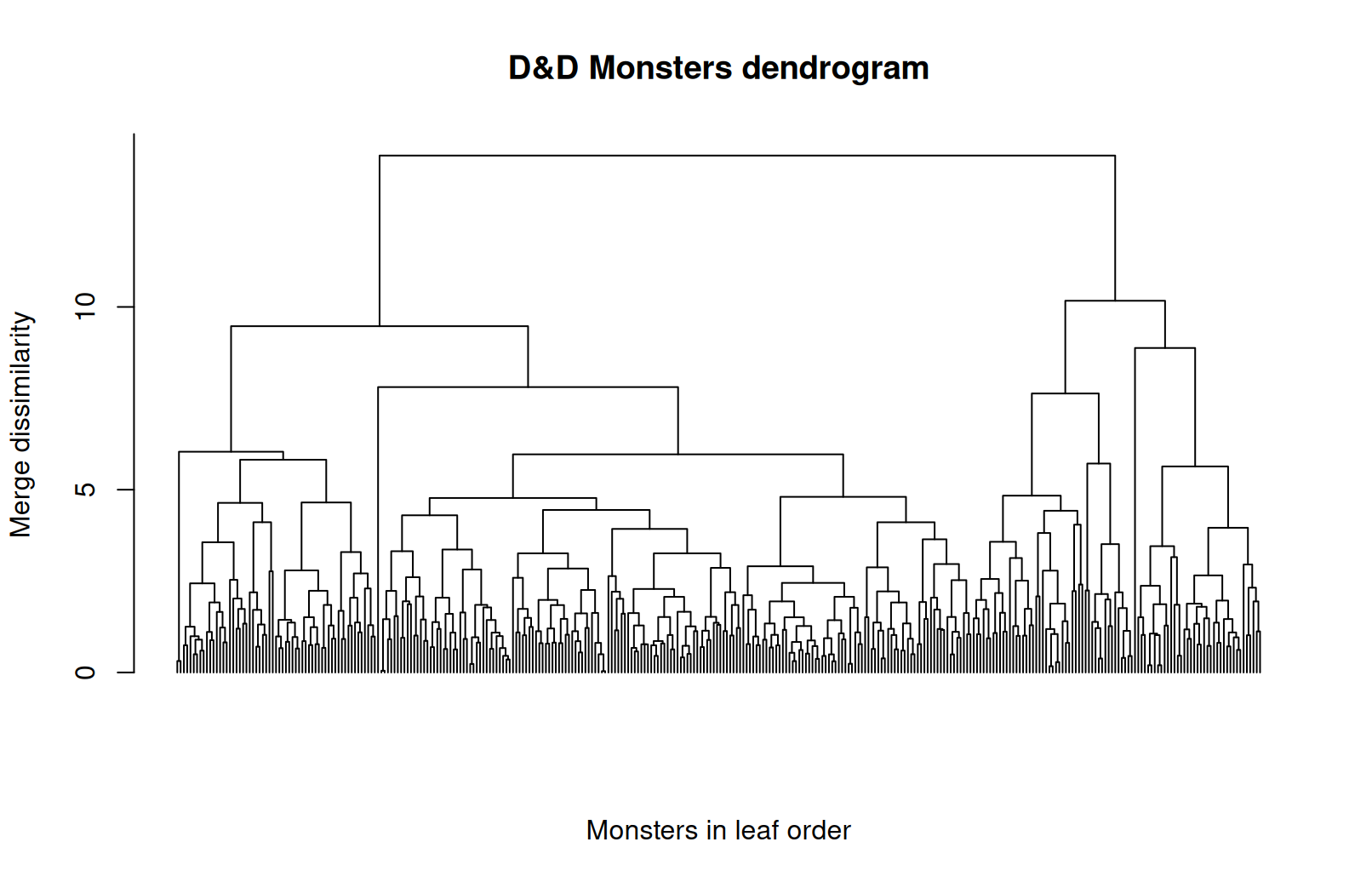
- The leaves on the x-axis are the individual monsters in the tree order.
- The height on the y-axis is the dissimilarity at which two clusters merge.
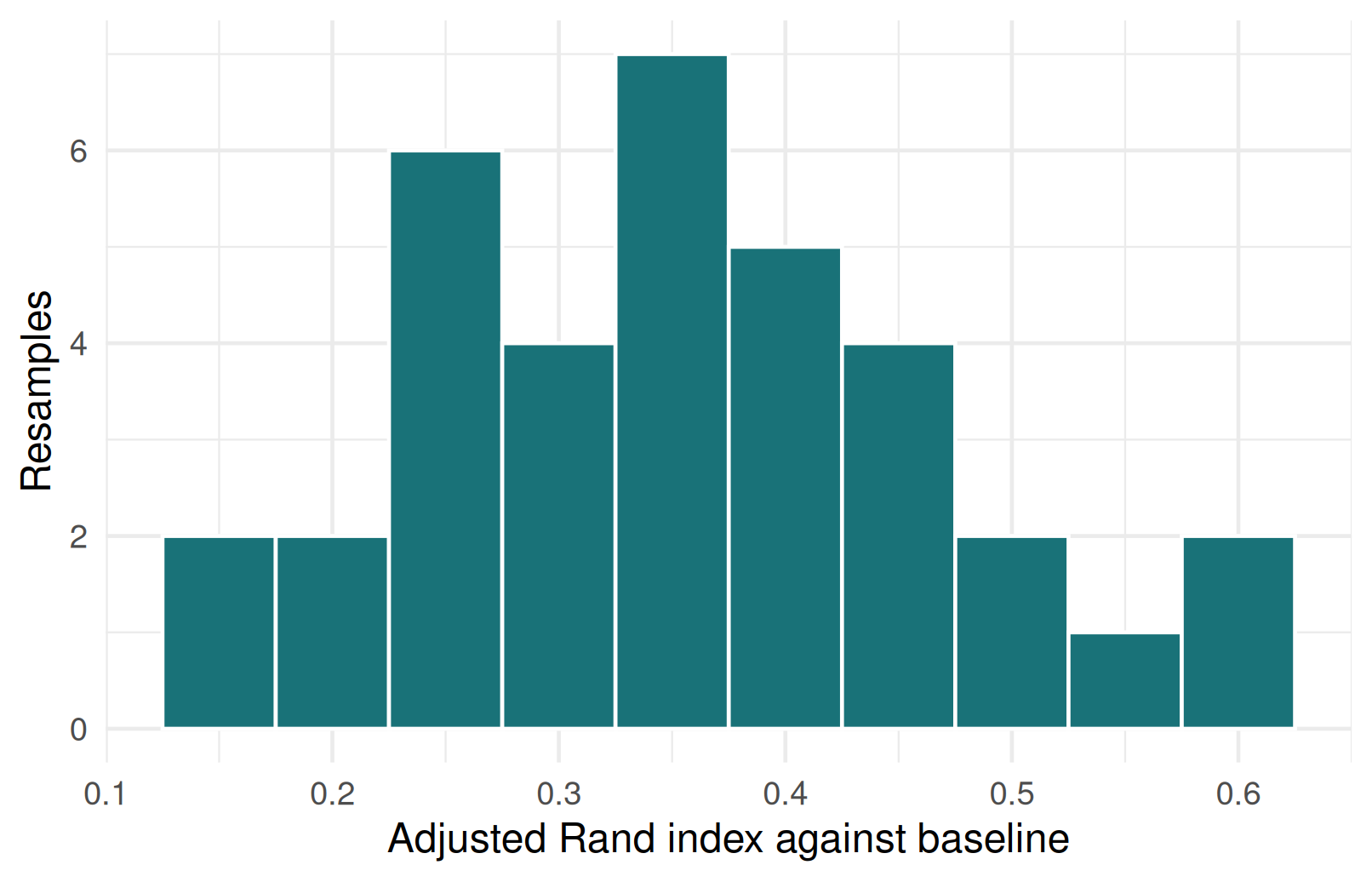
Bootstrap stability on monsters
- A bootstrap sample is a new dataset made by sampling the monsters with replacement from the original data.
- The tibble below contains one adjusted Rand index (ARI) value per bootstrap resample; here we used
35resamples. - The adjusted Rand index compares every pair of monsters across two partitions: did that pair stay together, or stay apart, in both clusterings? It then subtracts the agreement expected by chance, \[
\mathrm{ARI} = \frac{\sum_{ij}\binom{n_{ij}}{2} - E}{\frac{1}{2}\left[\sum_i \binom{a_i}{2} + \sum_j \binom{b_j}{2}\right] - E},
\] so
1means a perfect match to the baseline clustering,0means about as much agreement as random assignment, and negative values mean worse than random agreement. Higher values mean more stable cluster structure.
When K-means and PAM disagree
| Agreement | Count | Percent |
|---|---|---|
| Same cluster after relabelling | 233 | 70.6% |
| Different cluster | 97 | 29.4% |
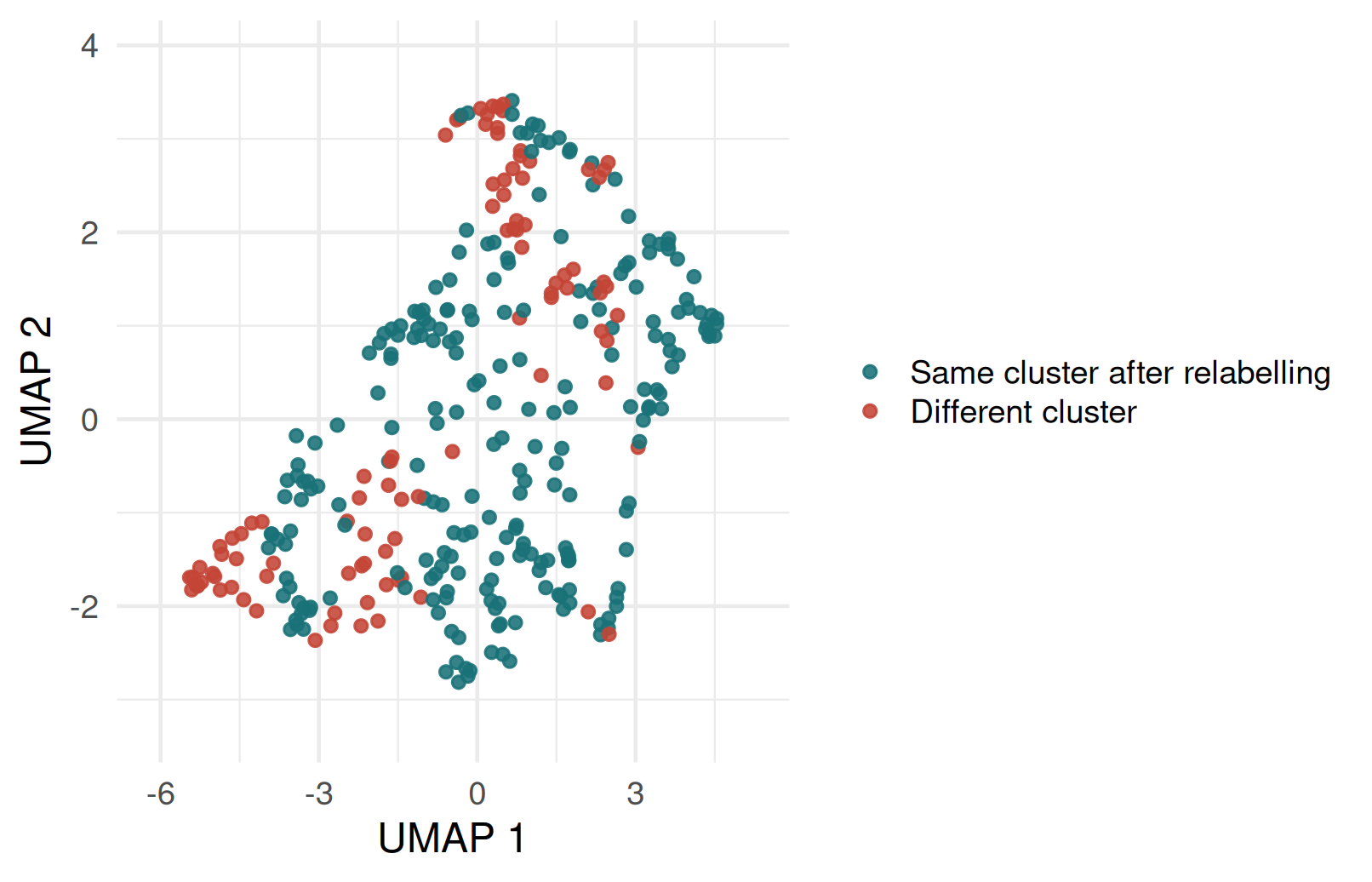
- Disagreement usually shows up around boundary observations or points affected by outliers.
- K-means is faster and works well for compact numeric clusters.
- PAM is more robust when the center should be a real observation or when outliers matter.
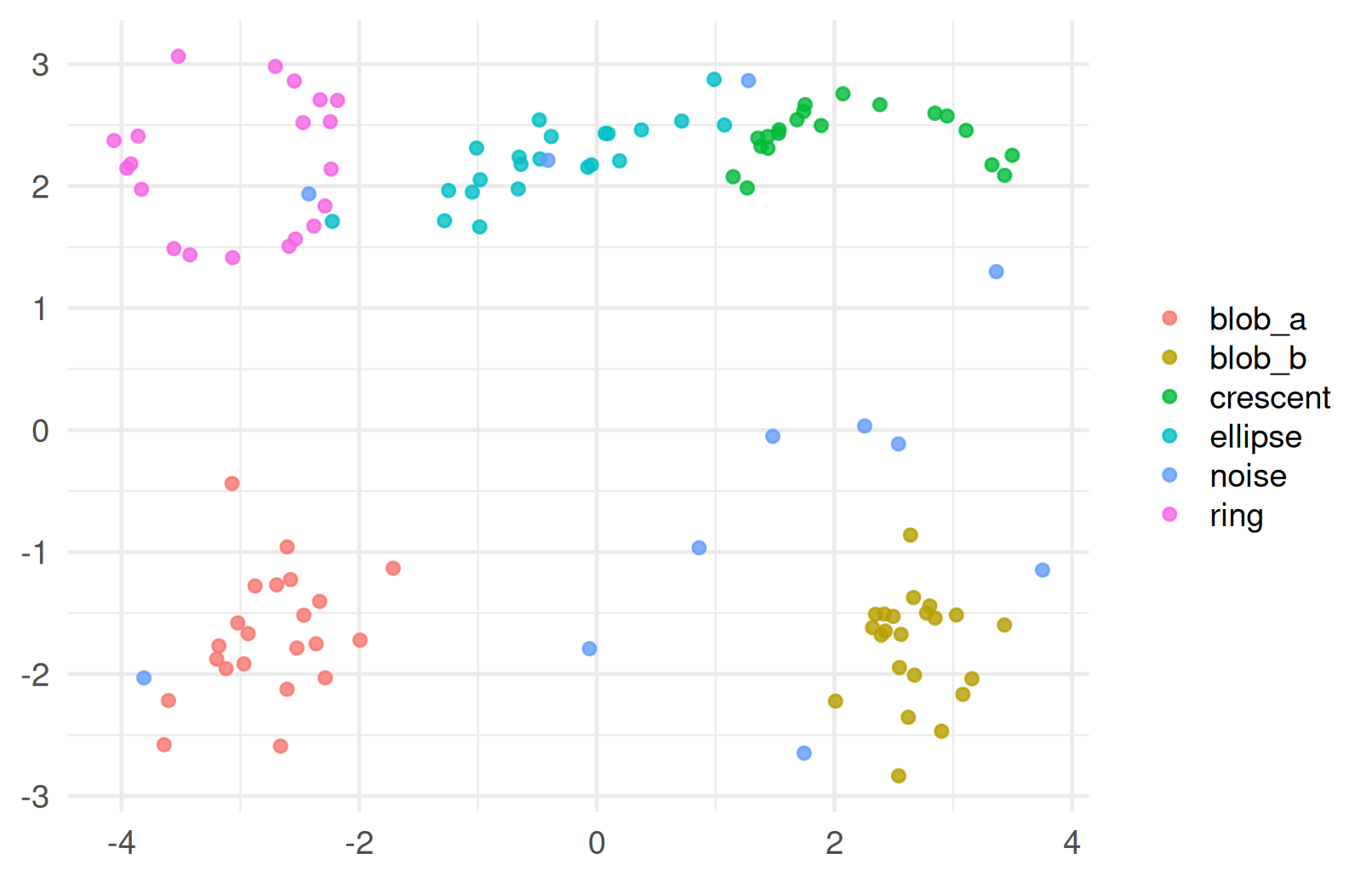
DBSCAN on our ring/crescent data
- This is exactly the kind of geometry where centroid-based methods tend to struggle.
- The ring and crescent are useful because they show how density-based clustering can recover non-convex shapes.
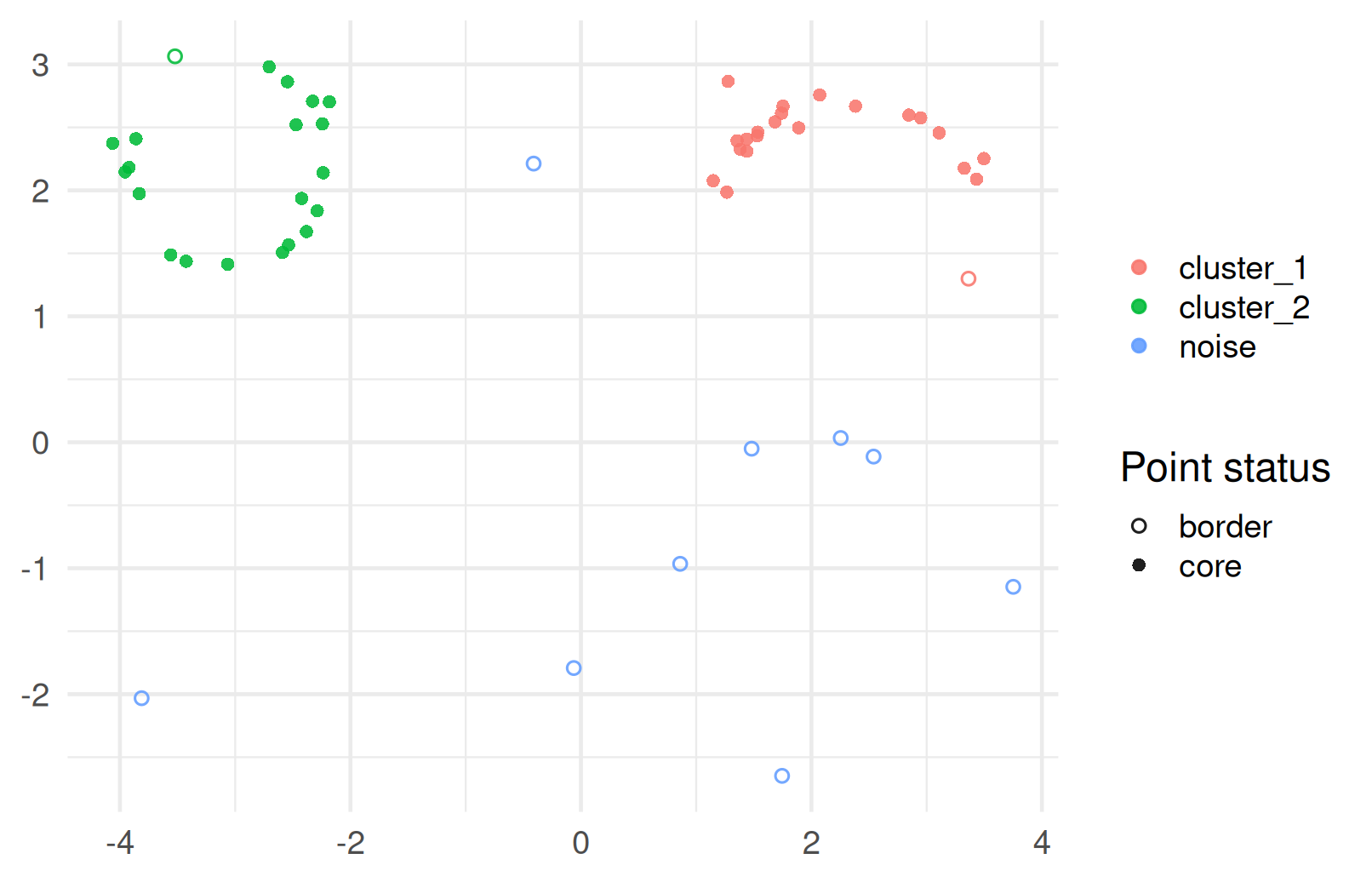
HDBSCAN
HDBSCAN stands for Hierarchical Density-Based Spatial Clustering of Applications with Noise.
- The “H” in HDBSCAN stands for “hierarchical,” which means it builds a hierarchy of clusters based on varying density thresholds.
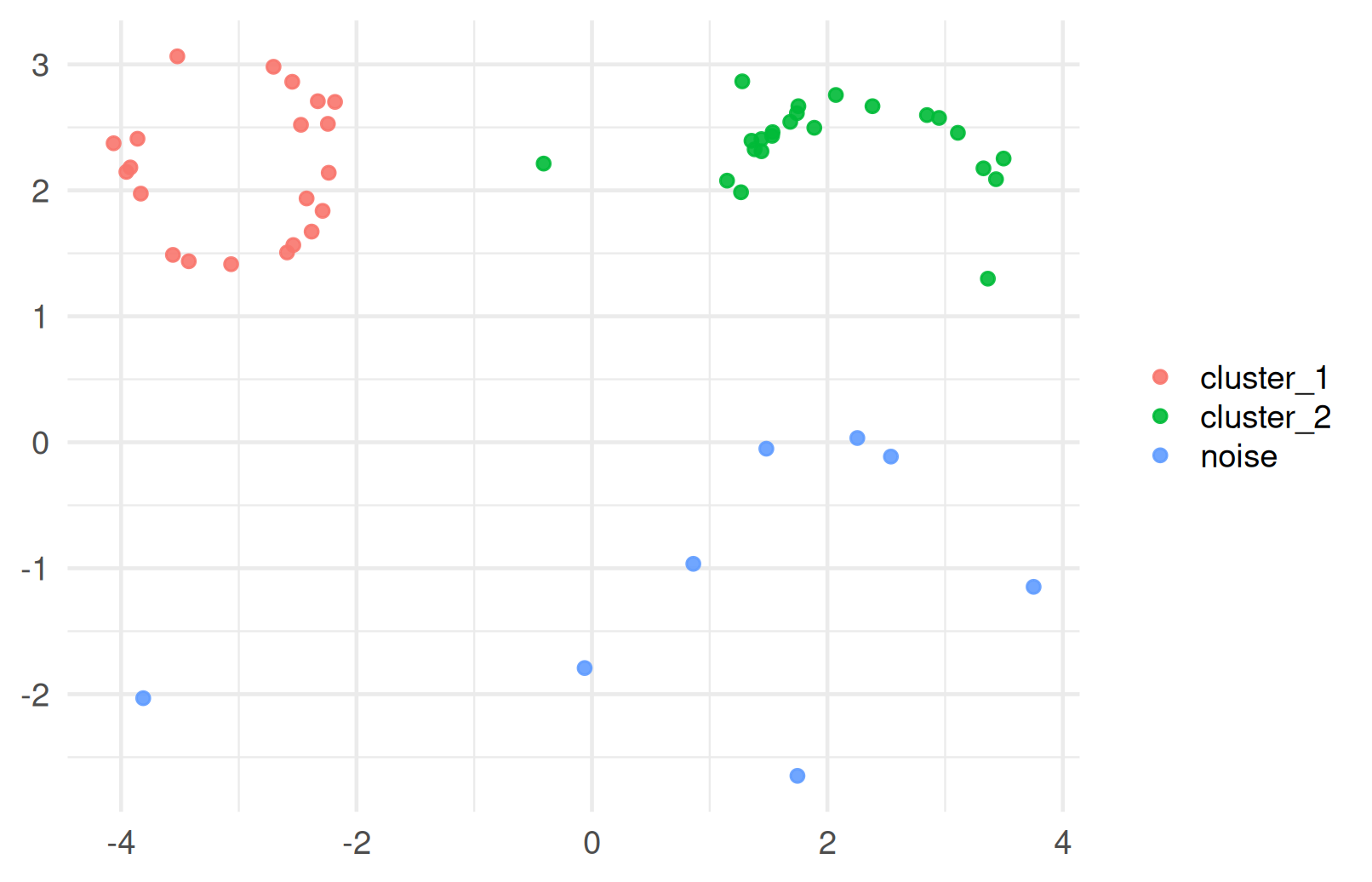
Advantages:
- Can handle clusters of varying densities better than DBSCAN.
- Provides a more robust clustering solution when the data contains clusters of different densities.
Disadvantages:
- More complex than DBSCAN and may require more computational resources.
- Like DBSCAN, it can be sensitive to parameter choices, although it has fewer parameters to tune.